
Given the increased demand for Mercator, we have moved it to a new faster infrastructure on www.plabipd.de/portal. This will support a greater number of concurrent jobs and will reduce the queueing time.
The results produces are identical but users can now submit larger jobs (this may be increased further pending a further upgrade).
Mercator Tutorial
Mercator is a tool to batch classify protein or gene sequences into MapMan functional plant categories. Many MapMan categories deal with metabolic pathways and enzyme functions, therefore using this pipeline a draft metabolic network can be established, especially after manual corretion of the automatically derived classification see e.g. May et al. 2008.
For a sequence clasification Mercator performs:
- Blast searches (searching for similarity to individual proteins/genes)
- Arabidopsis TAIR 10
- Large Parts of plant proteins from swiss-prot
- Uniref90
- RPS-Blast searches (searching for similarity to scoring matrices derived from similar proteins)
- and an InterPro scan
The results of the individual searches are then weighted by reliability. E.g. Uniref gets a low reliability since all proteins from Uniref90 are only classified based on keywords. The classifications with the highest reliabilty are retained. Current statistics (December 08):
- Accuracy ca. 90%
- Domains and families > 1000
- Proteins and genes > 30.000
To start a MapMan cluster job click "Create New Job".
Step 1 Sequence upload and option setup
You will be presented with an interface asking you for:

- JobName: Is a unique identifier given to your job. You can later use it to access your job.
- Name: is a name for this search for the users' convienience e.g. myprotein
- Email: is needed to alert you when the job is done. Depending on the selected options, the job can take quite some time.
- TAIR: check this if you want to blast your sequences against the Arabidopsis genome
- PPAP: check this if you want to blast your sequences against a large subset of the Swissprot plat proteins. (Arabidopsis is excluded from this set)
- UniRef: check this if you want to blast your sequences against the Uniref90 set. These are clusters of >=90% sequence identity. Please note, that Uniref Clusters are not classified into MapMan bins, rather, keywords are extracted from their description lines, thus this does not add much to the classification accuracy, but might be useful for annotating sequences see below
- Blast-Cutoff: This is the minimum bitscore value, below which blast hits are discarded. We suggest a value of 50.
- KOG: performs a RPS-blast search versus the KOG database
- CDD: performs a RPS-blast search versus the KOG database
- IPR: performs a full interproscan, comprising searches for Pfam,
- MULTIPLE: allows mutiple classifications of a protein
- PARANOID: Treats unknowns as a separate class which is competing for an annotation, this increases specificity at the cost of sensitivity
- ANNOTATE: adds the results of the blast and domain/protein family searches to the proteins or genes. This can be useful if the sequences are novel
- IS_DNA: if the sequence is a DNA sequence please check this box.
- LOAD_SEQUENCE: Please load a fasta formatted sequence file
Step 2 Check if everything is ok
Press "update job" to check if your submission is ok. You will get a small summary of your submission, in this case 22 sequences comprising 11361 nucleotides were submitted. If everything went ok, you will be presented with a new link: start Mercator
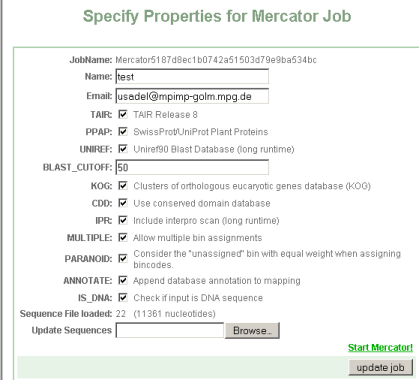
You will then be presented with a web site showing your sequence options, and the current status of your job.
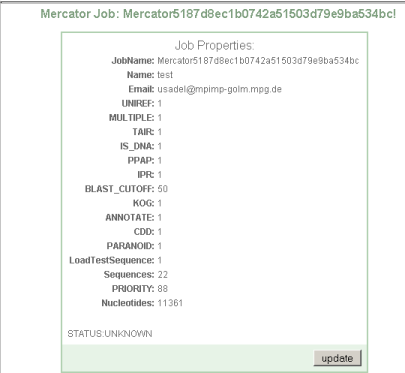
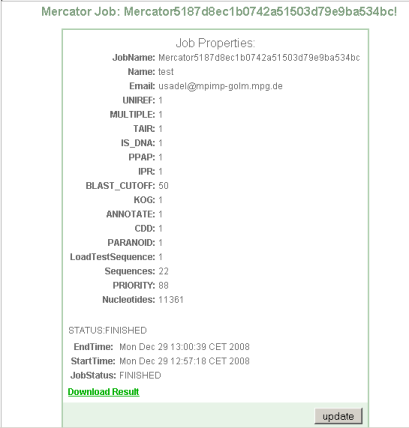
Step 3 Completion of batch job and download
Upon completion you will be offered a download link.
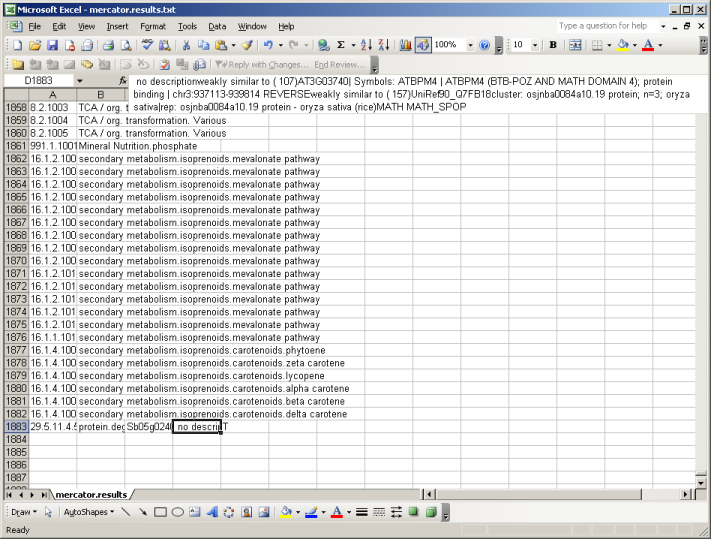
The file is available in zip format. After unziping the file you can open it in MapMan, in a text editor, or in MS Excel. If you open the file using excel, make sure that excel keeps everything formatted as text. (There are bins like 26.10 which get rounded or interpreted as dates by Excel sometimes)
At the top of the file you will be presented with all available MapMan Bins (classes). At the bottom you will find your sequences, grouped into Bincode and Name (the Classification into the MapMan system), an identifier and the description which has been extracted from the user-supplied FASTA file and enriched by the user checked searches. E.g. weakly similar to ( 107) AT3G03740| Symbols: ATBPM4 | ATBPM4 (BTB-POZ AND MATH DOMAIN 4); Here the number in brackets (107) gives the bitscore. In the following example the bist hit versus Arabidopsis is colored green, the one against Uniref90 in blue and potential domain names in red. All your sequences will be classified as "T" for transcripts.
| BINCODE | BINNAME | IDENTIFIER | DESCRIPTION | TYPE |
| 1.1 | PS.lightreaction | EST12345 | no description weakly similar to ( 107)AT3G03740| Symbols: ATBPM4 | ATBPM4 (BTB-POZ AND MATH DOMAIN 4); protein binding | chr3:937113-939814 REVERSE weakly similar to ( 157)UniRef90_Q7FB18cluster: osjnba0084a10.19 protein; n=3; oryza sativa|rep: osjnba0084a10.19 protein - oryza sativa (rice) MATH MATH_SPOP | T |
References
May, P., Wienkoop, S., Kempa, S., Usadel, B., Christian, N., Rupprecht, J., Weiss, J., Recuenco-Munoz, L., Ebenhöh, O., Weckwerth, W., Walther, D (2008) Metabolomics- and proteomics-assisted genome annotation and analysis of the draft metabolic network of Chlamydomonas reinhardtii. Genetics. 179:157-66.
Howell, K.A., Narsai, R., Carroll, A., Ivanova, A., Lohse, M., Usadel, B., Millar, A.H., Whelan, J. (200X) Mapping metabolic and transcript temporal switches during germination in Oryza sativa highlights specific transcription factors and the role of RNA instability in the germination process. Plant Physiol. accepted
Crowhurst, R.N., et al. (2008) Analysis of expressed sequence tags from Actinidia: applications of a cross species EST database for gene discovery in the areas of flavor, health, color and ripening. BMC Genomics. 9:351.